- Cos’è la fibrosi cistica?
- Quali sono le cause della fibrosi cistica?
- Sintomi iniziali
- Diagnosi di fibrosi cistica: neonati e adulti
- Quali sono i trattamenti per la fibrosi cistica?
- Si può prevenire?
Cos’è la fibrosi cistica?
↑ topLa fibrosi cistica è una malattia congenita geneticamente determinata che interessa più organi. È una patologia grave, con effetti dannosi sull’apparato respiratorio, l’apparato digerente, l’apparato endocrino e quello uro-genitale.
I pazienti affetti da fibrosi cistica presentano le vie respiratorie intasate da un muco denso e viscoso, difficile da espellere, che con il tempo rende sempre più difficile la respirazione. Le secrezioni polmonari, se non rimosse, possono ostruire le vie aeree, provocando anche la morte.
Inoltre, il muco che si concentra nei polmoni e nelle vie respiratorie costituisce un ambiente fertile per i batteri, con il conseguente rischio di contrarre infezioni polmonari, in particolare la polmonite.
Scopri la prestazione più adatta a te
Quali sono le cause della fibrosi cistica?
↑ topLa fibrosi cistica è una malattia genetica causata da un’alterazione del gene CFTR, che regola la produzione di una proteina fondamentale presente nelle cellule dei polmoni, del pancreas e di altri organi.
Questa proteina ha il compito di mantenere fluido il muco e di controllare la composizione del sudore. Nella fibrosi cistica, però, il suo funzionamento è compromesso, con conseguenze dirette sull’equilibrio dei fluidi dell’organismo.
Sintomi iniziali
↑ topI sintomi della fibrosi cistica variano a seconda dell’età in cui insorge la malattia, dei disturbi ad essa correlati e della disfunzione del gene CFTR. Due terzi dei casi di fibrosi cistica si manifestano entro il primo anno di vita. Una minoranza dei casi si manifesta in età adulta, cosa che rende la diagnosi particolarmente difficile.
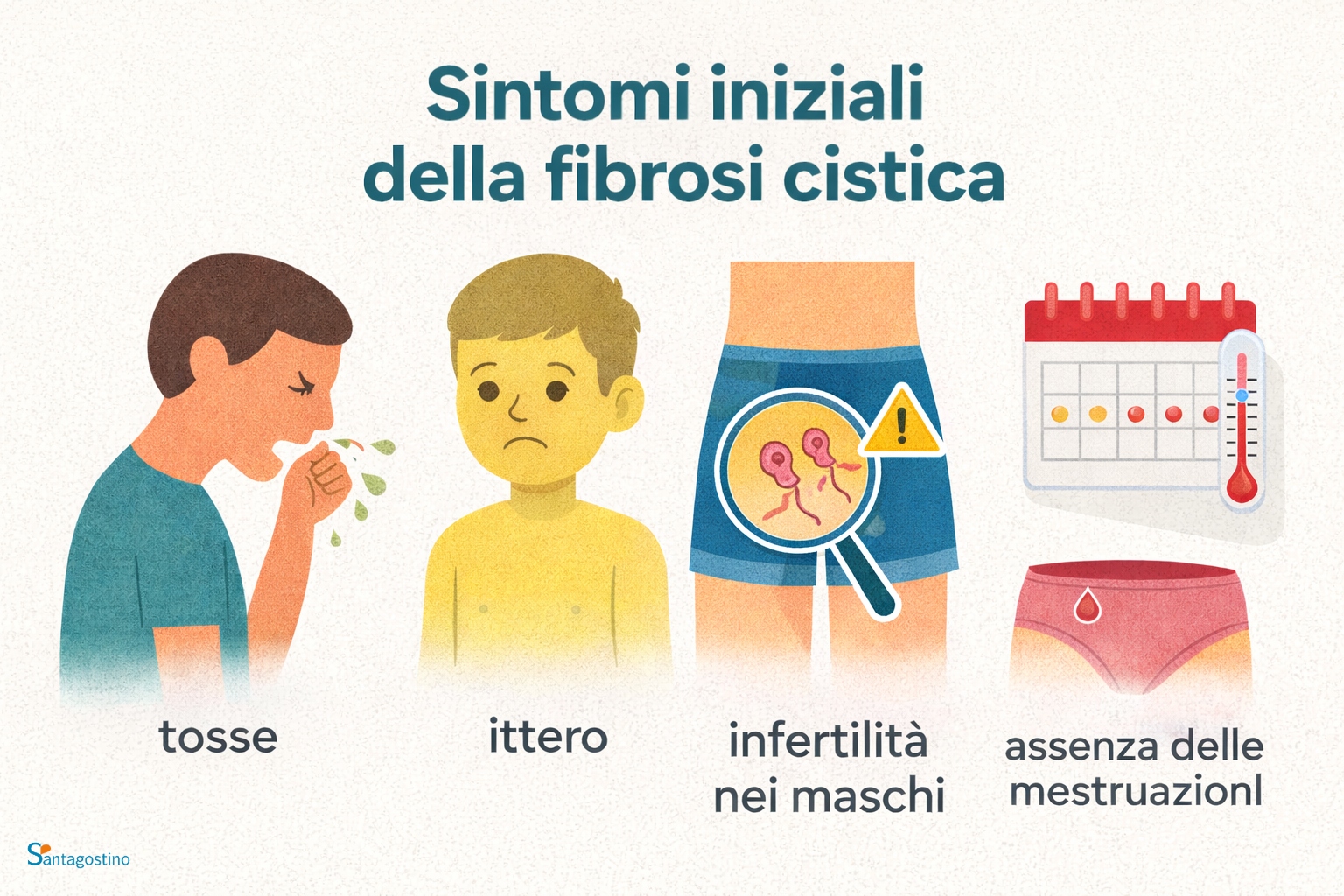
In generale, i segni e sintomi della malattia si presentano a carico del:
- sistema respiratorio: tosse con secrezioni dense e spesso purulente, polmoniti e sinusiti ricorrenti, sangue dal naso e dalla bocca
- apparato digerente: ittero, ostruzioni intestinali nel neonato o nel lattante, diarrea con feci maleodoranti e untuose, provocata dal malassorbimento intestinale,
- apparato uro-genitale: sviluppo sessuale secondario ritardato, infertilità dei maschi perché nascono senza i dotti deferenti, assenza delle mestruazioni per via della malnutrizione o delle ricorrenti infezioni respiratorie.
È prassi evitare che due malati di fibrosi cistica stiano in eccessivo contatto ravvicinato. Avendo una grande carica batterica a livello dei bronchi, la promiscuità tra malati può aggravare il quadro clinico di entrambi i pazienti. Non ci sono invece controindicazioni particolari nel caso di contatti con persone sane.
Scopri la prestazione più adatta a te
Diagnosi di fibrosi cistica: neonati e adulti
↑ topIl sospetto clinico di fibrosi cistica in un neonato si ha quando il piccolo non riesce a respirare a causa del muco che ostruisce le vie aeree e il pancreas non produce gli enzimi necessari alla digestione degli alimenti.
In presenza di casi di fibrosi cistica in famiglia o di sintomi sospetti, è consigliabile ricorrere ai test genetici e al test del sudore. Sono inoltre disponibili esami per la diagnosi prenatale e neonatale. È importante, infine, effettuare controlli mirati per valutare il coinvolgimento dei diversi organi e monitorarne l’evoluzione nel tempo.
Quando la malattia insorge in età adulta, la sua presenza è segnalata soprattutto da un’insufficienza pancreatica. Questa si manifesta con:
- cattiva digestione
- malassorbimento
- feci grasse, abbondanti e disfatte
- peso corporeo basso nonostante un’alimentazione adeguata.
L’insorgenza della malattia in età adulta è indice di minore gravità, perché il difetto genetico che la causa è meno importante. Di conseguenza, i sintomi sono più lievi e tardivi, e le persone tendono a vivere più a lungo.
Quali sono i trattamenti per la fibrosi cistica?
↑ topLa cura per la fibrosi cistica mira a prevenire il deterioramento della funzione respiratoria e a garantire una corretta nutrizione, nonostante il malassorbimento degli alimenti.
Tra i trattamenti più indicati, vi sono:
- fisioterapia respiratoria
- utilizzo di farmaci antibiotici, broncodilatatori, corticosteroidi
- somministrazione degli enzimi pancreatici a ogni pasto
- dieta equilibrata e ipercalorica
- disostruzione delle vie aeree
- iniezioni di insulina, nei pazienti che sviluppano il diabete
- trapianto di polmone, in caso d’insufficienza respiratoria grave, e fegato.
Esistono anche due farmaci che migliorano la funzione del gene CFTR, come lumacaftor e ivacaftor, efficaci solo in casi selezionati legati a specifiche mutazioni e non utilizzati come terapia standard.
Chi soffre di fibrosi cistica può avere figli?
↑ topLa fibrosi cistica colpisce purtroppo anche il sistema uro-genitale, con conseguenze sulla fertilità, soprattutto negli uomini.
| Infertilità maschile | La maggioranza degli uomini affetti da fibrosi cistica nasce senza i dotti deferenti, ovvero i canali che trasportano lo sperma dai testicoli. Questo significa che il loro sperma non è presente nel liquido seminale. Tale condizione, chiamata azoospermia ostruttiva, causa infertilità. Anche in assenza dei dotti, molti uomini producono comunque spermatozoi e possono ricorrere alla procreazione medicalmente assistita per avere figli. |
| Problemi riproduttivi femminili | Anche se la fibrosi cistica non causa infertilità nelle donne come negli uomini, può comunque creare ostacoli al concepimento. La malattia può:
Tuttavia, anche i problemi di fertilità femminile possono essere affrontati con le moderne tecniche di inseminazione e fecondazione assistita. È importante ricordare che, essendo una malattia ereditaria, i test genetici sono necessari per valutare il rischio di fibrosi cistica nei figli, a seguito del trattamento dell’infertilità. |
Aspettative di vita
↑ topL’aspettativa di vita per chi soffre di questa patologia è molto migliorata negli ultimi anni.
Purtroppo, però, la malattia rimane ancora seria e la sua gravità varia da persona a persona, a seconda di come si sviluppano le complicazioni polmonari e altri disturbi associati.
In generale, possiamo dire che l’aspettativa di vita per i pazienti è di circa 40 anni.
Scopri la prestazione più adatta a te
Si può prevenire?
↑ topÈ possibile prevenire il rischio di avere un bambino affetto da fibrosi cistica attraverso l’analisi del proprio DNA.
Durante la gravidanza, è possibile eseguire esami genetici tramite amniocentesi o villocentesi, per determinare se il neonato è sano o affetto da patologia.